SCRAM
SCRAM is lightweight Python package for aligning small RNA reads to one
or more reference sequences and producing publication-quality images.
Developed by Stephen Fletcher @ the laboratory of Prof. Bernie Carroll, University of Queensland
Installation
Scram is written in Python 3.5. Install scram and its dependencies with pip:
pip install scram
Or download and extract the tarball, then run:
python setup.py install
Input File Format
Reference File : DNA nucleotides only (AGCT) - FASTA format
Sequence File : Collapsed (unique) reads - DNA nucleotides only (AGCT) - FASTA format
Post-processing of FASTQ reads to collapsed FASTA format can be carried out using the
FASTX-Toolkit from the Hannon Lab.
Collapsed reads are unique, and contain the read count in the header.
An example of the required sequence file format:
head seq1.fa
>1-607041
TCGGACCAGGCATCATTCCCC
>2-202886
TCGGACCAGGCTTCATACCCC
>3-71446
TCCCAAATATAGACAAAGCA
Usage
scram analysis_type reference_file [-h] [-s1 [SEQ_FILE_1 [SEQ_FILE_1 ...]]]
[-s2 [SEQ_FILE_2 [SEQ_FILE_2 ...]]] [-nt SRNA_LEN] [-f FILE_NAME]
[-p PROCESSES] [-min_read MIN_READ_SIZE]
[-max_read MAX_READ_SIZE] [-min_count MIN_READ_COUNT]
[-win SMOOTH_WIN_SIZE] [-ylim YLIM] [-no_csv] [-no_display]
[-split] [-pub] [-V]
Analysis types
den: align reads of a single sRNA class (eg. 21 nt) from one or more replicate sequence files to a single reference sequence (-s1and-ntrequired)mnt3dm: align 21, 22 and 24 nt reads from one or more replicate sequence files to a single reference sequence (-s1required)CDP: count aligned reads of a single sRNA class (eg. 21 nt) to multiple reference sequences. Counts for two sets of one or more replicate sequence files are plotted as (x,y) coordinates for each reference (-s1,-s2and-ntrequired)CDP_single: count aligned reads of a single sRNA class (eg. 21 nt) to multiple reference sequences. Counts are written to a .csv file with a column for each read file. No plot output. (-s1and-ntrequired)
-h: Help message-s1: Sequence file 1 (if more than 1 replicate sequence file, read counts are averaged)-s2: Sequence file 2 (if more than 1 replicate sequence file, read counts are averaged)-nt: sRNA length to analyse-p: number of processes (CPU cores) to use in CDP analyses (default=4)-f: Figure output file name (if not auto-generated). Use 'auto' to auto-generate-min_read: Minimum length of sRNA reads used for normalisation (default=18)-max_read: Maximum length of sRNA reads used for normalisation (default=32)-min_count: Minimum read count for an sRNA to be aligned and used for normalisation (default=1)-win: Window size for smoothing of den plots (default=50, min = 6)-ylim: +/- y-axis limit on plots-no_display: Do not display plot on screen-no_csv: Do not generate the .csv alignment file-split: Split read alignment counts based on no. of alignments (i.e. if a read aligned in 2 positions, the read count at each position is halved)-pub: Remove all axis labels and legends for preperation of figures for publication-V: Show program's version number and exit
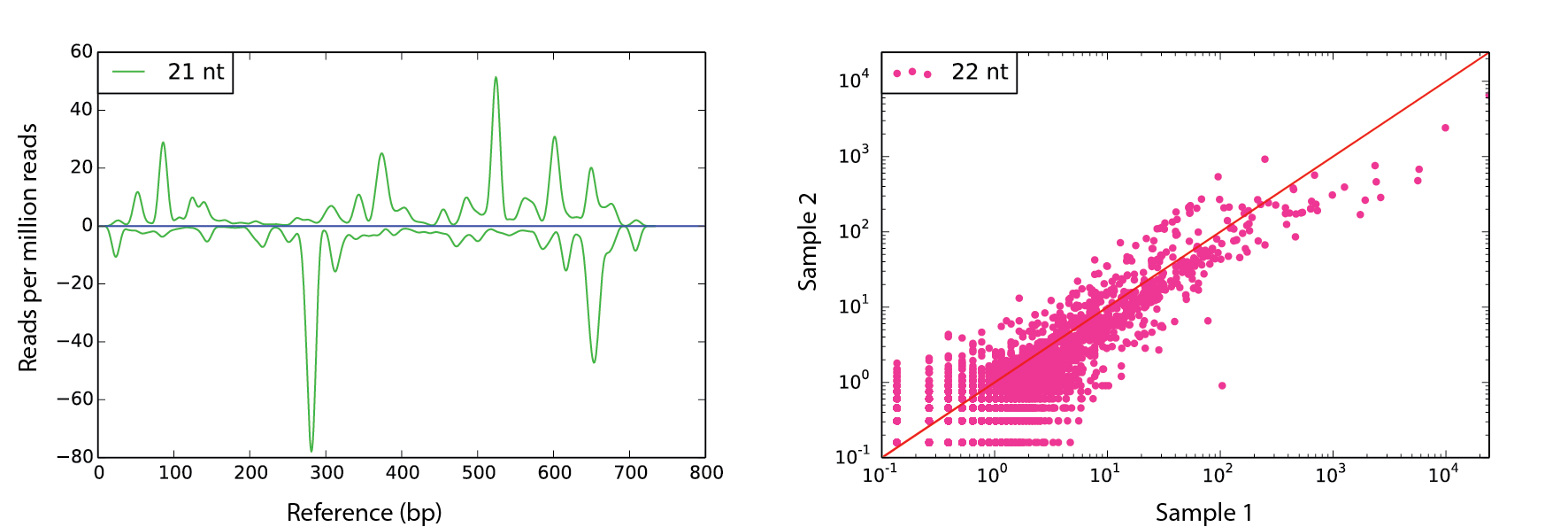
den Example
scram den ./ref.fa -s1 seq1.fa -nt 24 -win 30 -f fig1.pdf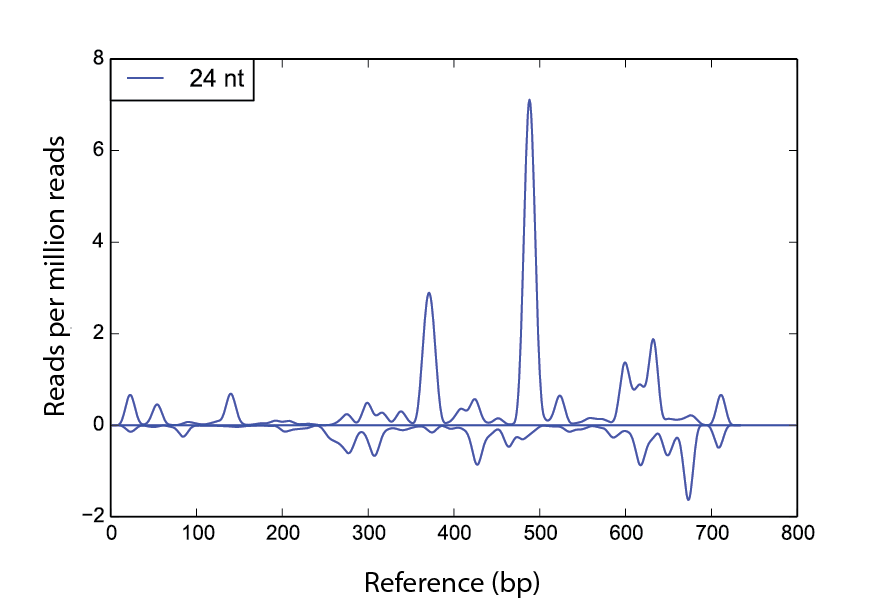
mnt3dm Example
scram mnt3dm ./ref.fa -s1 seq1.fa -win 20 -ylim 110 -f fig2.pdf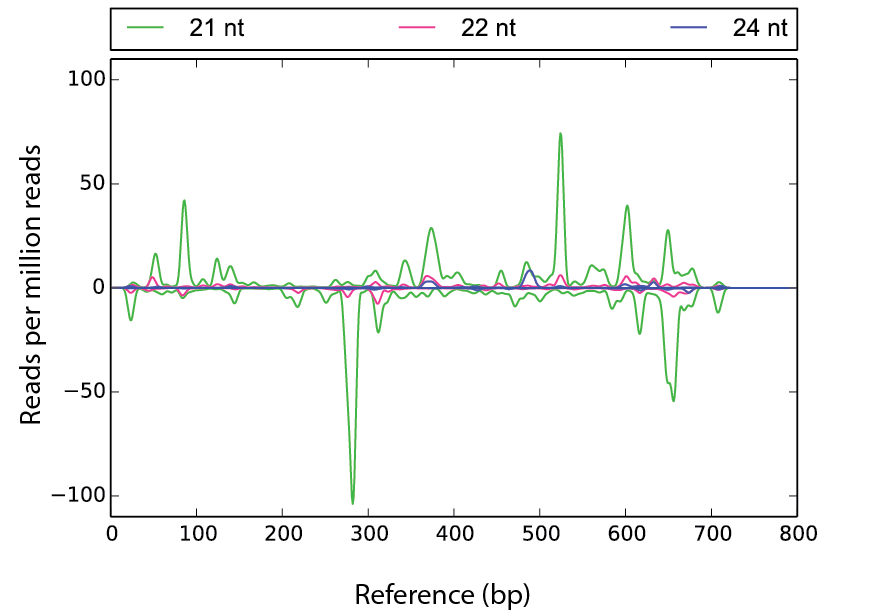
CDP Example
scram CDP ./cDNAs.fa -s1 seq1.fa -s2 seq2.fa -nt 21 -f fig3.pdf -split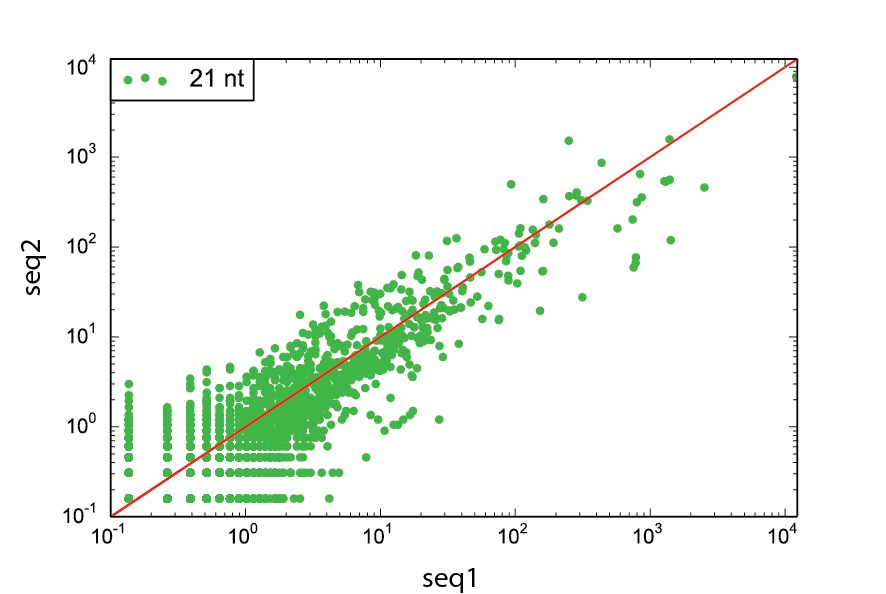
(c) 2016 - Stephen Fletcher. MIT License